 BWT-1(y#Lrabtooar) of Computational Biology
BWT-1(y#Lrabtooar) of Computational Biology
We apply techniques from computer science, maths and stats -- Statistical inference, information theory, Data structures and Algorithms, Combinatorial Optimisation etc. -- to address key computational challenges that arise in biological data, predominantly in those involving protein 3D structures and 1D sequences.
Some open-source programs, web utilities and resources developed at LCB
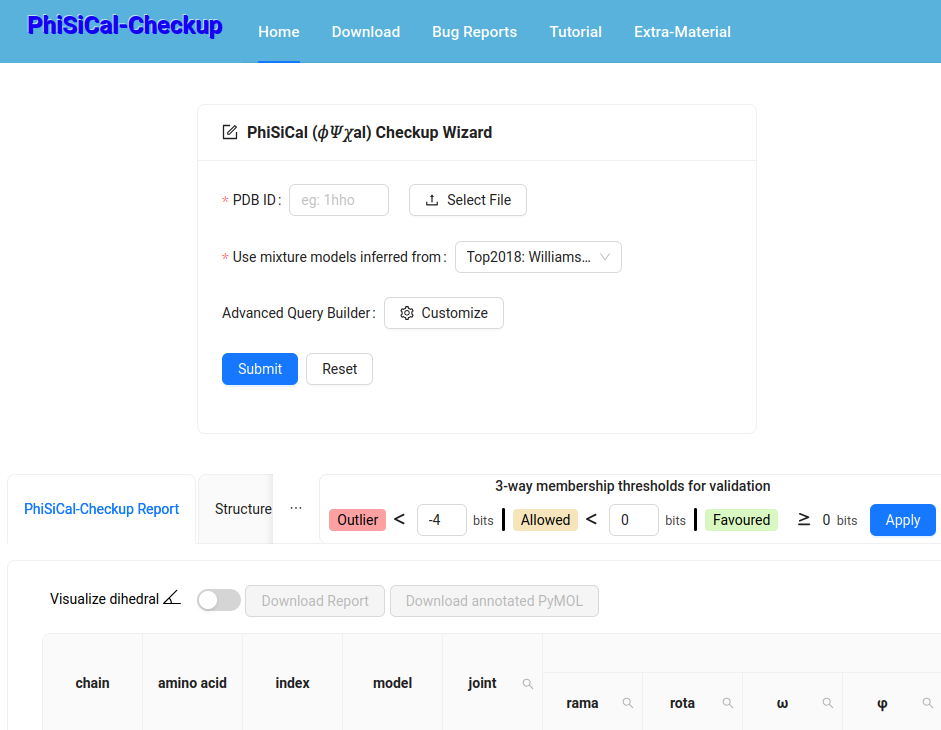
This framework/software/web-server supports reliable validation of experimental 3D protein coordinates using PhiSiCal (𝜙𝛹𝜒al) mixture models that provide amino acid-specific {joint,marginal,conditional} probability distritutions. (PNAS 122(1):e2416301121 2025)
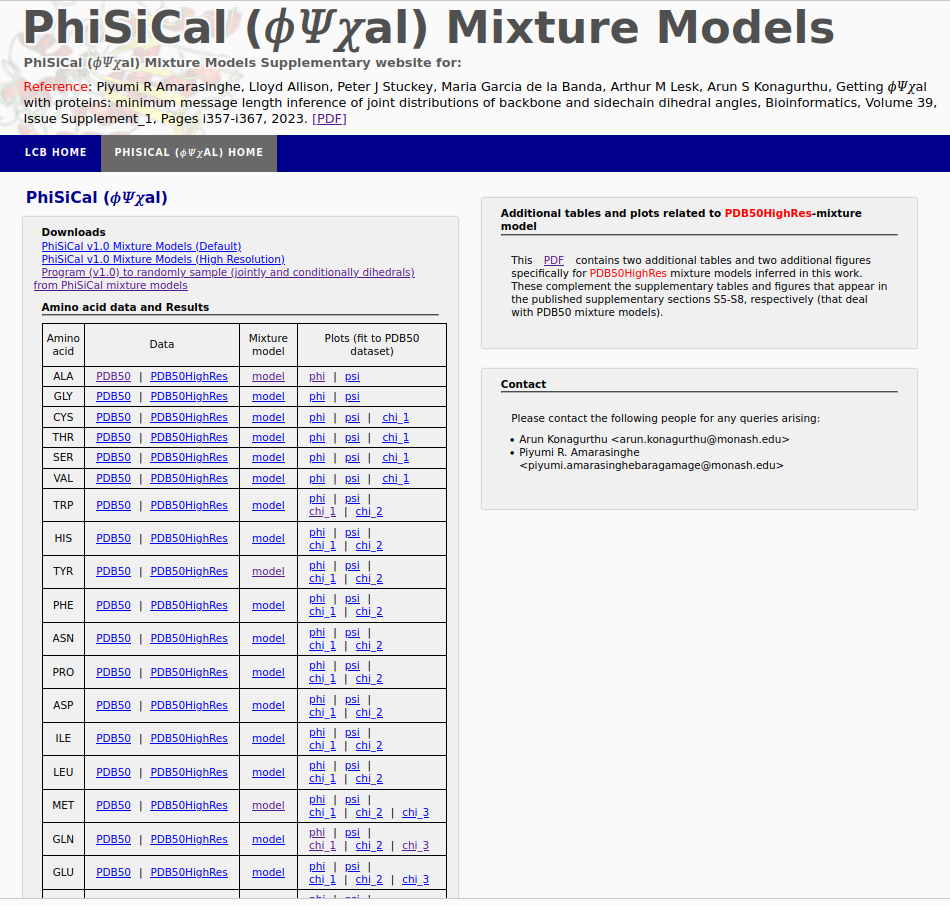
PhiSiCal (𝜙𝛹𝜒al) mixture models are Minimum Message Length (MML) inferred statistical models exlaining the joint distribution of backbone (𝜙,𝛹) and sidechain (𝜒) dihedral angles of individual amino acids, supporting accurate conformation sampling and many other applications to protein studies.
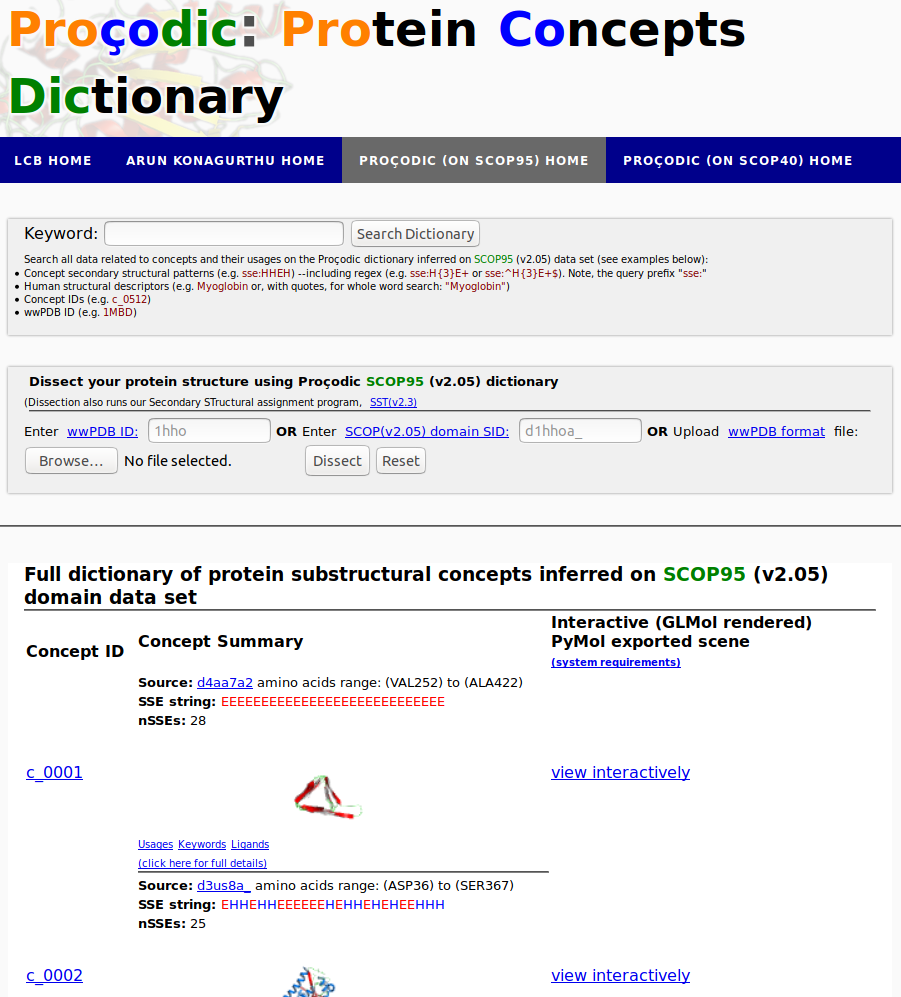
Proçodic is an interactive web server capturing the dictionary of topologically conserved secondary structures (a.k.a concepts), that forms the architectural 'basis set' of the observered universe of protein structures.

MMLigner is a command-line program and webserver to infer pairwise protein 3D structure alignments and also identify closely-competing alignments. It uses the MML-based statistical inference framework supported by probability distributions on 3D spheres.
MMLigner
seqMMLigner is a command line program to infer alignments between amino acid SEQUENCES under the MML framework.
Dinithi won the 2019 Ian Lawson Van Toch Memorial Award (Outstanding Student Paper) for this work.

SST is a web server to assign secondary structure to protein coordinate data using the Bayesian method of Minimum Message Length inference. Identifies helices, turns of various types, and strands of a sheet.

MUSTANG is a command line program to produce multiple structural alignments given the three-dimensional coordinates of proteins.
MMLSUM is a 'time' parameterised Stochastic Markov model of amino acid substitutions, inferred using MML along with its companion 'time' parameterised Dirichlet distributions modelling the alignment three-state machine. Together they form a complete set of models for amino acid substitutions, insertions and deletions.
Super is a web server and a program to rapidly screen the entire (up-to-date) PDB and identify similar oligopeptide fragments. The method mathematically guarantees to find all superposable fragments for a given query that fits within a user-prescribed threshold of root-mean-squared deviation (RMSD).
3Dlib
Superpose3D is a C++ library that supports least-squares superposition of 3D vector sets. This library implements sufficient statistics for this superposition problem, and allows updating existing superpositions (under vector set addition and symmetric difference) in constant time.
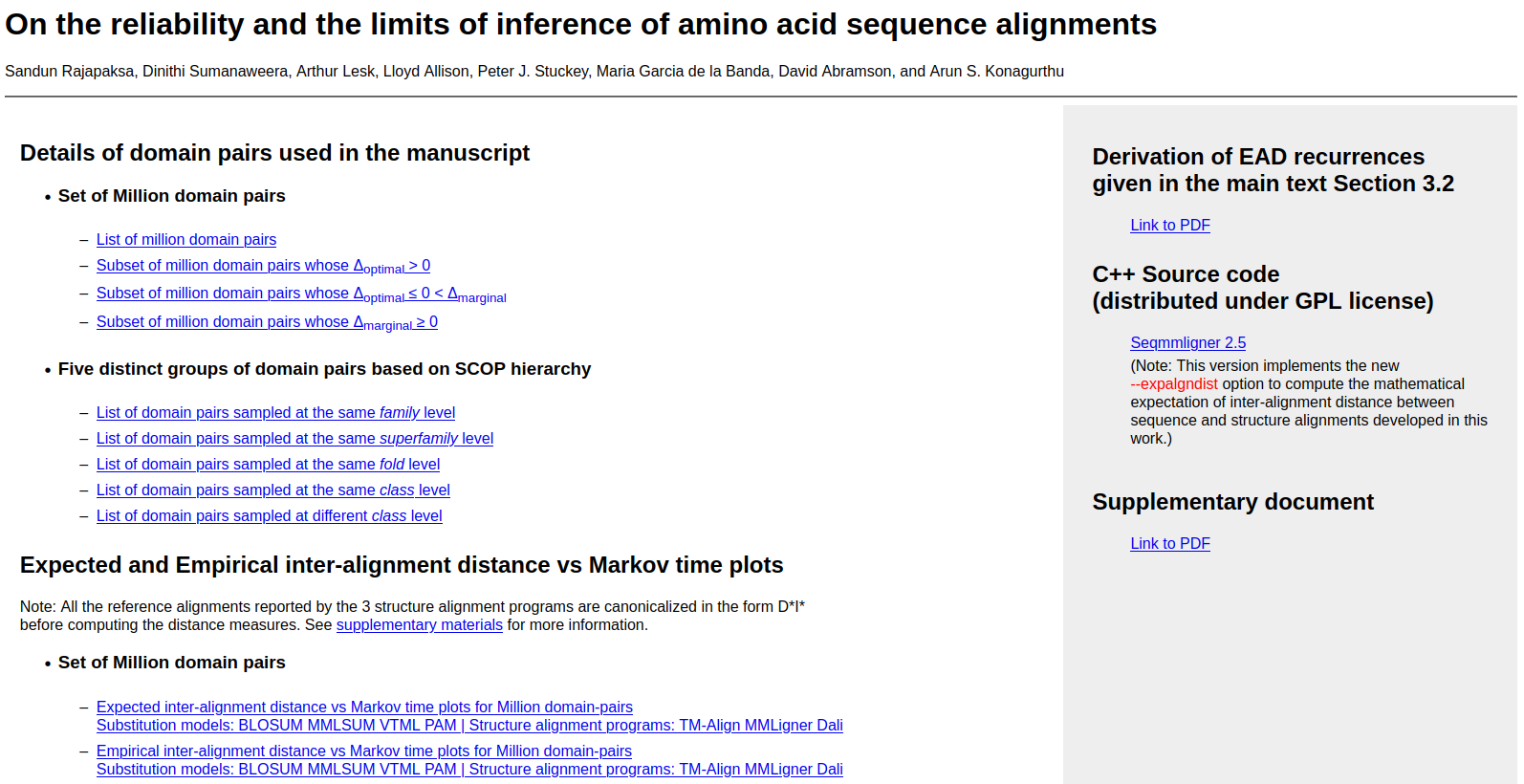
EAD (Expected Alignment Distance) builds on the seqMMLigner framework to estimate the (mathematically strict) expectation of inter-alignment distance between the relationships decipherable from 1D amino acid sequence information and 3D structure information.
Team
- David Abramson (@Univ.Qld.)
- Lloyd Allison
- Maria Garcia de la Banda
- Arun Konagurthu (Research lead)
- Arthur M. Lesk (@Penn State)
- Peter Stuckey
- Piyumi R. Amarasinghe (Baragamage) (PhD student; Feb 2022-)
- ↪ First author on PNAS 2025 paper as part of her PhD research (click)
Past students
- Sandun Rajapaksa (PhD student; 2019-2023) [Thesis]
- Dinithi Sumanaweera (PhD student; 2017-2021) [Thesis]
- ↪ Winner 2019 Ian Lawson Van Toch Memorial Outstanding Student Paper Award [photo])
- ↪ Won 2021 Mollie Holman Award for the Best Thesis
- ↪ Won 2021 Marie Sklodowska-Curie postdoctoral fellowship of European Commission (S. Teichmann's lab at Univ. of Cambridge and EMBL)
- James Collier (PhD student; 2012-2016; VC's commendation for PhD thesis excellence 2016) [Thesis]
- Parthan Kasarapu (PhD student; 2012-2016; FIT Best Doctoral Paper Award 2016) [Thesis]
{kind=link}
Past research staff
- Gitansh Khirbat (Research Assistant on ARC DP150100894 Grant; 2017-2018)
- Ramanan Subramanian (Research Fellow on ARC DP150100894 Grant; 2015-2017)